
Dr. Alessandro Giardini
An aortopulmonary window is a rare and serious congenital heart defect, but it is also one that responds very well to timely treatment. Most parents have never heard the term before their baby is diagnosed, and the experience of learning about it, often in the first days or weeks of a child's life, can be frightening and disorienting.
This page explains clearly what an aortopulmonary window is, why it causes problems, what the warning signs are, how it is diagnosed and treated, and what the outlook is after surgery. Dr. Alessandro Giardini's specialist work in complex congenital heart disease at Great Ormond Street Hospital means that families dealing with this diagnosis can access focused, experienced care.
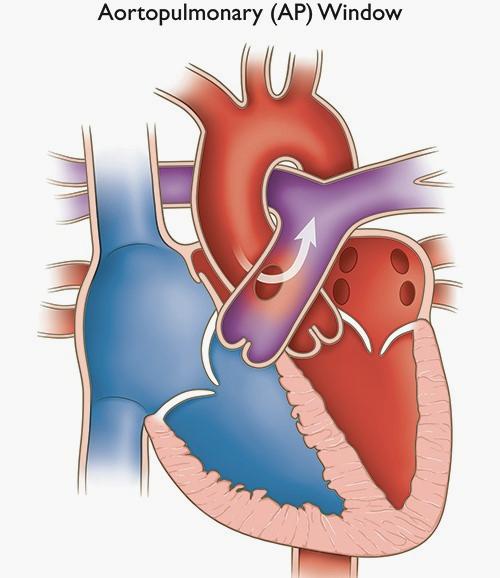
An aortopulmonary window (also called an AP window, an aortopulmonary septal defect, or a common arterial window) is an abnormal opening between the aorta and the main pulmonary artery. These two great arteries normally leave the heart as completely separate vessels: the aorta carries oxygen-rich blood from the heart to the body, and the pulmonary artery carries oxygen-poor blood from the heart to the lungs. In a baby with an aortopulmonary window, there is a hole or gap in the wall that should separate these two vessels.
Because pressure in the aorta is much higher than in the pulmonary artery, blood is pushed continuously from the aorta through the opening into the pulmonary artery. The result is that too much blood flows to the lungs, overloading them and the left side of the heart. This causes breathlessness, heart failure, and, if the condition is untreated for any length of time, progressive high pressure in the lung blood vessels (pulmonary hypertension).
The key distinction from truncus arteriosus is worth making clearly, because parents and even some referring clinicians sometimes confuse the two. In an aortopulmonary window, the aorta and pulmonary artery are distinct vessels with their own valves (an aortic valve and a pulmonary valve), but an abnormal connection exists between them. In truncus arteriosus, there is a single shared vessel leaving the heart with a single valve. They are different conditions, with different surgical approaches and different implications.
Aortopulmonary window is very rare, accounting for less than 0.5 per cent of all congenital heart defects. In absolute numbers, fewer than one baby in ten thousand is born with the condition. Because it is so rare, it is important that children with this diagnosis receive care from a congenital heart team with specific experience in the condition.
Aortopulmonary windows come in three main types, classified according to the position of the opening between the two arteries.
Type I (proximal) is the most common. The opening sits close to the base of the two vessels, just above the valves. This type accounts for the majority of cases.
Type II (distal) involves the opening sitting further away from the valves, closer to where the right pulmonary artery branches off. This type is less common and can be more technically complex to repair because of its proximity to the right pulmonary artery origin.
Type III (combined or total) describes a large defect involving most of the shared wall between the two vessels.
For most parents, the precise type matters less than the size of the defect and whether other cardiac anomalies are present alongside it. Surgeons use the classification to plan the specific repair technique.
A particularly rare and complex variant is sometimes called Berry syndrome, in which an aortopulmonary window occurs alongside an interrupted aortic arch and abnormal origin of the right pulmonary artery from the aorta. This combination requires highly specialised surgical management.
Not always, and this is an important point for families to understand. Approximately 50 per cent of children with an aortopulmonary window have at least one other congenital heart defect alongside it. Common associated conditions include interrupted aortic arch (a break or severe narrowing in the aorta, which must be repaired simultaneously), coarctation of the aorta (a narrowing of the aorta), ventricular septal defect (a hole between the lower chambers), atrial septal defect, tetralogy of Fallot, transposition of the great arteries, and abnormalities of the coronary arteries.
The presence of associated defects affects surgical planning, operative complexity, and the timeline for recovery. Contemporary surgical series show that even children with complex associated lesions can have excellent outcomes at specialist centres.
An aortopulmonary window develops early in fetal life, during the embryological process in which the single great arterial trunk that exits the developing heart normally divides into two separate vessels: the aorta and the pulmonary artery. When this division is incomplete, an opening persists between the two vessels. The condition is congenital, meaning it is present from birth, and is not caused by anything a parent did or did not do during pregnancy.
Most cases occur in isolation without an identified genetic cause. Associated chromosomal abnormalities or syndromes are less common than with some other congenital heart defects, though a thorough genetic assessment forms part of the overall workup.
Most babies with a significant aortopulmonary window develop symptoms within the first days or weeks of life, and the bigger the opening, the earlier and more pronounced those symptoms tend to be. This is because a large opening allows an enormous amount of blood to flood into the lungs, rapidly overwhelming the heart's capacity.
Common signs include fast or laboured breathing (tachypnoea), which may be noticed by parents as unusually rapid chest movements or grunting. Feeding is typically difficult: babies tire quickly during feeds, sweat during them, and take in too little milk to gain weight adequately. Poor weight gain, unusual pallor, persistent irritability, and recurrent chest infections are all reported. A heart murmur is almost always audible on examination.
In the small number of cases where the opening is very small, symptoms may be minimal and the condition found incidentally. In most cases, however, the defect is large, symptoms appear quickly, and urgent investigation is needed.
Parents should know that if a large aortopulmonary window is left untreated for a long time, it can lead to irreversible damage of the lung blood vessels (pulmonary vascular disease) and eventually to Eisenmenger syndrome, a condition in which the lung pressures become so high that blood flow reverses direction and the child becomes blue (cyanosed). Once Eisenmenger syndrome develops, the defect can no longer be closed surgically. This is one of the central reasons that early diagnosis and prompt repair are so important.
Sometimes, yes. An aortopulmonary window can occasionally be identified on foetal echocardiography, either during the routine 20-week anomaly scan or on a specialist fetal cardiac assessment. However, prenatal diagnosis is difficult and is often missed. The abnormal connection between the two great arteries can be subtle on prenatal imaging, and many babies receive their diagnosis only after birth when symptoms appear or a murmur is heard.
Antenatal diagnosis, when it does occur, allows delivery planning at or near a specialist cardiac centre, which enables immediate assessment and treatment without delay.
The echocardiogram is the cornerstone of diagnosis. A skilled operator can visualise the abnormal connection between the aorta and the pulmonary artery and assess its size, position, and relationship to surrounding structures including the coronary arteries and pulmonary artery branches. The echocardiogram also evaluates ventricular function, pulmonary artery pressures, and any associated defects.
The ECG typically shows right ventricular hypertrophy or combined ventricular hypertrophy, reflecting the increased work placed on the heart by the large left-to-right shunt.
A chest X-ray usually shows an enlarged heart (cardiomegaly) and increased prominence of the pulmonary blood vessels, consistent with high pulmonary blood flow.
Routine newborn pulse oximetry measures the oxygen level in the blood. Low or borderline readings in a newborn prompt further cardiac investigation.
CT angiography provides detailed three-dimensional images of the great arteries, pulmonary artery branches, aortic arch, and coronary arteries. It is particularly valuable for defining complex anatomy before surgery, especially when associated defects such as interrupted aortic arch or abnormal pulmonary artery origins are present.
Cardiac MRI defines anatomy and cardiac function without radiation, and is useful in older children where greater anatomical detail is needed without the radiation burden of CT.
Cardiac catheterisation, in which a fine tube directly measures pressures inside the heart and great vessels, is not routinely needed in newborns where the diagnosis is clear on echocardiography and CT. It is used selectively: in babies presenting later in infancy (beyond six months of age) when pulmonary hypertension may already be established, and in older children presenting late, to assess pulmonary vascular resistance and determine whether the defect remains operable. In rare cases with very small defects, catheterisation may also be used therapeutically (see below).
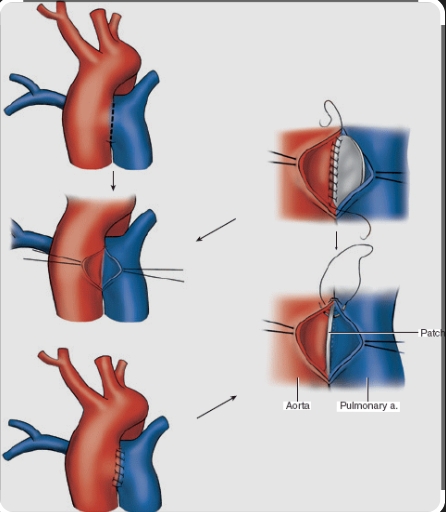
Surgical closure is the definitive treatment for the vast majority of children with an aortopulmonary window. The operation takes place on cardiopulmonary bypass (the heart-lung machine), which temporarily supports the circulation while the surgeon works on the heart and great arteries.
The most common approach involves opening the aorta and closing the window from the inside using a patch of fabric or pericardium. This transaortic patch repair prevents any risk of damage to the pulmonary artery from the repair itself. A double-patch technique is sometimes used, placing patches on both the aortic and pulmonary artery sides of the defect. The correct approach depends on the type of the window and the associated anatomy.
If associated defects such as interrupted aortic arch or VSD are present, surgeons aim to repair everything in a single operation where possible, avoiding the need to expose the baby to multiple bypass runs.
Contemporary surgical results are excellent. A 25-year series from Texas Children's Hospital (2024) reported a median operative age of 19 days, with only a single mortality across 29 patients including nine with interrupted aortic arch. Multiple large series confirm that early outcomes after AP window repair approach zero mortality for isolated defects.
Surgery needs to happen as soon as possible after diagnosis. Waiting allows the pulmonary hypertension to progress. Repair performed before irreversible pulmonary vascular changes develop produces the best long-term outcomes. In practice, most babies with an aortopulmonary window undergo surgery within days to weeks of diagnosis.
For a small minority of children with small, restrictive defects, transcatheter (catheter-based) closure using a device delivered through a blood vessel represents an alternative to open surgery. This is not the standard approach and applies only to carefully selected anatomies, but it is worth knowing that this option exists. The decision requires specialist cardiac catheterisation expertise.

Before surgery, babies may need support with medications to manage heart failure, including diuretics to reduce fluid congestion and in some cases medications to reduce pulmonary arterial pressure. Respiratory support may also be needed. The goal is to stabilise the baby optimally before the operation.
Following repair, babies move to the cardiac intensive care unit, where they are closely monitored by a specialised team. A breathing tube, chest drains, and monitoring lines are normal in the immediate post-operative period. These are gradually removed as recovery progresses. Most babies remain in hospital for one to three weeks after surgery, depending on the complexity of the repair and whether associated defects were present.
Following discharge, babies need close follow-up with a paediatric cardiologist. The heart takes time to adapt to the new, normalised circulation, and echocardiographic review ensures recovery is proceeding well.
Babies under one month of age who have undergone complex congenital heart surgery should receive neurodevelopmental screening before the age of two, as there is a recognised association between complex neonatal cardiac surgery and neurodevelopmental outcomes that benefit from early identification and support.
The outlook after prompt surgical repair of an aortopulmonary window is generally very good. For isolated AP windows repaired early in infancy, before significant pulmonary vascular disease has developed, contemporary series report mortality approaching zero and excellent long-term cardiac function. Most children grow up to lead active, normal lives without ongoing cardiac limitations.
The long-term prognosis depends primarily on two factors: the presence and complexity of associated cardiac defects, and whether pulmonary vascular disease had time to develop before repair. Children with isolated AP windows repaired promptly do best. Those with complex associated lesions, particularly interrupted aortic arch, have a more variable course, and a small proportion need further interventions (such as balloon dilatation of the aortic arch or pulmonary arteries) as they grow.
All children who have had an aortopulmonary window repaired need lifelong specialist cardiac follow-up. This is a routine, planned programme of review rather than a reflection of ongoing instability, and in most children the reviews become less frequent as time passes and the heart demonstrates stable long-term function.
For those who had late repair or in whom pulmonary hypertension was already present at the time of surgery, ongoing monitoring of lung pressures and exercise capacity forms an important part of long-term follow-up. Young women with repaired AP window need specialist pre-conception counselling before pregnancy.
If your baby has been diagnosed with an aortopulmonary window, urgent paediatric cardiology and cardiac surgery input at a specialist congenital heart centre is needed. This is not a condition where watchful waiting is appropriate.
A specialist review is also valuable for families seeking a second opinion on diagnosis or the surgical plan, for families who want expert assessment of the anatomy before consenting to surgery, or for older children or young adults with repaired AP window who need ongoing congenital heart disease follow-up.
An aortopulmonary window is a rare congenital heart defect in which there is an abnormal opening between the aorta (the large artery carrying blood to the body) and the pulmonary artery (the large artery carrying blood to the lungs). Because aortic pressure is higher, blood flows continuously through the opening into the lungs, causing flooding of the lung circulation and heart failure.
Yes. A significant aortopulmonary window causes heart failure and, if untreated, leads to progressive and eventually irreversible lung vessel damage (pulmonary hypertension and Eisenmenger syndrome). Prompt diagnosis and surgery are essential. With timely repair, however, most children do very well.
The most common symptoms in the first days and weeks of life are fast or laboured breathing, difficulty feeding, excessive sweating during feeds, poor weight gain, unusual tiredness, and recurrent chest infections. A heart murmur is almost always present on examination.
A significant AP window always needs closure. In rare cases where the defect is very small and restrictive, catheter-based closure may be an option, but in most babies open-heart surgical repair is the appropriate treatment.
Surgery is performed as soon as possible after diagnosis, typically within days to weeks of birth. The longer a significant defect remains open, the greater the risk of irreversible lung vessel damage. Contemporary series report median operative ages of around 19 to 22 days.
Sometimes. An AP window may be identified on foetal echocardiography, but prenatal diagnosis is often difficult and many cases are found only after birth. When a prenatal diagnosis is made, it allows delivery to be planned at a specialist cardiac centre.
No. In an aortopulmonary window, the aorta and pulmonary artery are separate vessels with separate valves, but an abnormal hole connects them. In truncus arteriosus, there is a single common arterial trunk leaving the heart. They require different surgical approaches and have different implications.
In approximately 50 per cent of cases, yes. Common associated defects include interrupted aortic arch, coarctation of the aorta, ventricular septal defect, and tetralogy of Fallot. Associated defects are repaired at the same time as the AP window where possible.
For isolated AP windows repaired early in infancy, the outlook is excellent, with very low mortality in contemporary surgical series and most children growing up to live normal, active lives. The presence of complex associated defects or delayed diagnosis with established pulmonary hypertension affects outcomes and requires more careful long-term follow-up.
Yes. All children with repaired aortopulmonary window need ongoing specialist cardiac review, even after successful surgery. In most cases, follow-up becomes less frequent as the child demonstrates stable cardiac function over time.
Author: Dr. Alessandro Giardini, MD, PhD
Written 17/04/2026