Loeys-Dietz Syndrome in Children and Adolescents
Written by Dr Alessandro Giardini, Consultant Paediatric and Adolescent Cardiologist
What Is Loeys-Dietz Syndrome?
Loeys-Dietz syndrome (LDS) is a rare genetic condition that affects connective tissue, the material that gives strength and flexibility to blood vessels, bones, muscles, and skin. Two doctors, Bart Loeys and Harry Dietz, first identified it in 2005 at Johns Hopkins University in the United States.
Before this discovery, doctors often mistakenly diagnosed children with LDS as having Marfan syndrome, a better-known condition that can look quite similar. We now know that LDS is a separate condition with its own risks and its own approach to treatment.
LDS affects roughly 1 in 50,000 people. It can involve many parts of the body, but the biggest concern is the tendency to develop aneurysms: areas where the wall of an artery becomes weak and stretches outward like a balloon. These most commonly occur in the aorta, the main blood vessel that carries blood away from the heart, but they can happen in arteries anywhere in the body. If nobody finds and monitors an aneurysm, there is a risk that it could tear or burst, which is a medical emergency.
The reassuring news is that with early diagnosis, regular check-ups, medication, and surgery when needed, children and young people with Loeys-Dietz syndrome can live full, active lives. Doctors once told families that life expectancy might be limited, but the treatments available today have improved the outlook enormously. As a paediatric cardiologist at Great Ormond Street Hospital, I have seen children and families navigating this diagnosis regularly, and I know how important it is to have clear, practical information from the outset.
What Causes Loeys-Dietz Syndrome?
A change (known as a mutation) in one of several genes causes LDS. These genes all form part of a signalling system in the body called the TGF-beta pathway, which helps cells grow, develop, and maintain healthy tissues including the walls of blood vessels.
When one of these genes changes, the signalling system becomes overactive. This weakens connective tissue throughout the body and leads to the problems that characterise LDS.
Six known types of Loeys-Dietz syndrome exist, each linked to a different gene. Type 2 (caused by the TGFBR2 gene) is the most common, accounting for more than half of all cases. Types 1 and 2 together make up the large majority. Types 3 to 6 occur less frequently but doctors increasingly recognise them as genetic testing becomes more widely available.
LDS follows what doctors call an autosomal dominant pattern of inheritance. This means that having just one copy of the altered gene is enough to cause the condition. If a parent has LDS, each of their children has a 1 in 2 (50 per cent) chance of inheriting it.
However, in about 3 out of 4 cases, LDS happens for the first time in a child with no family history at all. The gene change occurs by chance during early development. This means that even if no one else in the family has had LDS, a child can still develop the condition.
One important thing for families to understand is that LDS varies enormously from person to person. Even two members of the same family carrying the exact same gene change can experience the condition very differently: one may have quite mild features while the other has more significant problems.
What Are the Symptoms of Loeys-Dietz Syndrome?
LDS can affect several parts of the body. Not every child will have all of these features, and their severity can range from very mild to more significant.
Heart and blood vessels
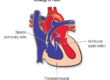
This is the most important area. Most people with LDS will develop some widening of the aorta, particularly at the aortic root (the section closest to the heart). Aneurysms can also develop in other arteries throughout the body, including those in the head, neck, chest, and abdomen.
A feature quite distinctive to LDS is arterial tortuosity so that the arteries take on a twisted, spiralling course rather than running straight. Doctors most commonly notice this in the neck vessels and, while it does not usually cause problems in itself, it provides an important clue that helps make the diagnosis.
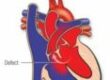
Some children with LDS are born with heart defects such as a bicuspid aortic valve (a valve with two flaps instead of the usual three), a hole in the heart (atrial or ventricular septal defect), or a patent ductus arteriosus. Doctors also sometimes find mitral valve prolapse (where the valve between the left chambers of the heart is slightly floppy).
Face and head
Children with LDS may have eyes that sit more widely apart than usual (a feature doctors call hypertelorism). The uvula, the small piece of tissue that hangs down at the back of the throat, may split in two (bifid uvula) or appear unusually broad. Some children have a cleft palate, and in some cases the bones of the skull fuse earlier than normal (craniosynostosis). These facial features tend to become less obvious as a child grows.
Bones and joints
Many children with LDS have very flexible joints (joint hypermobility). Other features can include long fingers and toes, chest wall shape differences such as a sunken chest (pectus excavatum) or a protruding chest (pectus carinatum), curvature of the spine (scoliosis), flat feet, and clubfoot. About 15 per cent of people with LDS have some instability in the bones at the top of the neck (cervical spine), and doctors need to check for this.
Skin
The skin may feel unusually soft or velvety, look thin, or bruise easily. Wounds may heal slowly and leave wide or unusual scars.
Allergies and gut problems
Around a third of people with LDS have significant food allergies. Asthma, eczema, and hay fever also occur more frequently than in the general population. Some children experience tummy problems including abdominal pain, difficulty absorbing food, or inflammation in the gut.
Eyes
Eye problems such as a squint (strabismus) may occur. However, dislocation of the lens of the eye, a hallmark of Marfan syndrome, does not happen in LDS. This is one of the key features that helps doctors tell the two conditions apart.
How Is Loeys-Dietz Syndrome Diagnosed?
Diagnosing LDS involves putting together information from a physical examination, heart and blood vessel scans, and genetic testing.
Physical examination
A specialist, usually a paediatric cardiologist and/or a clinical geneticist, carries out a thorough examination, looking for the characteristic features of LDS, including any differences in the face, skin, and joints. Because LDS shares features with Marfan syndrome and other connective tissue conditions, a cardiologist experienced with inherited cardiac conditions needs to assess the child carefully. In my practice, I frequently see children referred with a possible connective tissue disorder where the distinction between LDS, Marfan syndrome, and related conditions needs careful working through.
Echocardiogram (heart ultrasound)
This is usually the first heart test. It uses sound waves to create a picture of the heart, allowing the doctor to measure the size of the aorta and check how the heart valves work. It causes no pain and involves no radiation.
CT or MRI scan of the blood vessels
A detailed scan of the arteries from head to pelvis forms an essential part of the assessment. Unlike in Marfan syndrome, aneurysms in LDS can develop in arteries far from the heart and not just in the aorta. These scans also reveal whether the arteries follow the twisted, tortuous pattern that characterises LDS. Doctors often prefer MRI in children because it avoids radiation.
Genetic testing
A blood test can pinpoint the specific gene change responsible. This confirms the diagnosis and also tells doctors which type of LDS a child has. Genetic testing matters not just for the child but for the wider family. It allows parents, brothers, and sisters to undergo testing so that anyone else who carries the gene change can start surveillance before any problems develop.
Other tests
Depending on the child’s individual features, the doctor may also arrange X-rays of the neck to check for cervical spine instability, a skull scan if early fusion of the skull bones raises concern, and an eye examination.
If a child receives a diagnosis of LDS, both parents should ideally have genetic testing (or at least a clinical examination) to find out whether the gene change came from a parent or occurred for the first time in the child.
What Treatments Are Available?
No cure for Loeys-Dietz syndrome currently exists, but effective management significantly reduces the risk of serious complications and allows children and young adults to thrive. With the right care, the outlook for children diagnosed today is far better than it was even a decade ago.
Medication
The main aim of drug treatment is to lower blood pressure and reduce the strain on the walls of the arteries, slowing down any tendency for the aorta or other arteries to widen.
Doctors commonly prescribe beta-blockers to slow the heart rate and lower blood pressure. They also widely use losartan, a medication from a group called angiotensin receptor blockers (ARBs). Losartan holds particular interest in LDS because, as well as lowering blood pressure, it appears to act on the very signalling pathway (TGF-beta) that drives the condition. In some cases where the aorta continues to enlarge despite losartan treatment, doctors may try a related medication called irbesartan.
Regular monitoring
Ongoing surveillance forms one of the most important parts of managing LDS. Most children have an echocardiogram (heart ultrasound) every 6 to 12 months to track the size of the aorta and check heart valve function. Doctors usually arrange a full CT or MRI scan of the arteries from head to pelvis once a year to look for aneurysms elsewhere in the body.
The exact schedule of monitoring depends on each child’s individual findings and how stable their condition remains over time. I work with each family to develop a surveillance plan tailored to their child, balancing thoroughness with keeping the burden of hospital visits manageable.
Surgery
When an aneurysm reaches a certain size, doctors recommend planned (preventive) surgery to avoid the risk of a tear or rupture. In LDS, the point at which surgeons advise operating comes earlier than in Marfan syndrome, because tears can happen when the aorta is still relatively small, typically around 4.0 cm in teenagers and adults, or sooner in younger children if the aorta is growing quickly.
The most common operation is a valve-sparing aortic root replacement. The surgeon removes the weakened section of the aorta and replaces it with a synthetic tube while keeping the patient’s own aortic valve. This well-established procedure carries an excellent safety record. Surgeons may also need to operate at other sites if aneurysms develop in other arteries.
Exercise and activity
Children with LDS should stay active. Gentle to moderate activities such as swimming, cycling, walking, hiking, and recreational tennis are generally safe. A useful rule of thumb is that your child should feel comfortable enough to hold a conversation while doing the activity. As a specialist with a particular interest in exercise physiology in children with heart conditions, I help families find the right balance between staying active and staying safe as both are important.
Families should steer children away from competitive and contact sports, heavy weight-lifting, and intense exercises like push-ups, sit-ups, and pull-ups, as these activities cause sudden increases in blood pressure that put extra stress on the arteries.
Bone and joint care
An orthopaedic specialist may get involved if a child has scoliosis, clubfoot, flat feet, or chest wall differences. Physiotherapy can help with joint hypermobility.
Allergy and gut management
The appropriate specialists should help manage food allergies, asthma, and gut problems. It is worth noting that children with LDS who experience migraines should generally avoid triptan medications (such as sumatriptan), as these narrow blood vessels and doctors do not consider them safe in connective tissue disorders.
Team approach
Because LDS affects many body systems, care works best when a team of specialists coordinates it. This may include a paediatric cardiologist, cardiac surgeon, clinical geneticist, orthopaedic surgeon, eye specialist, allergy specialist, and gastroenterologist. A lead clinician who oversees the big picture helps ensure nothing falls through the gaps.
Frequently Asked Questions
How is Loeys-Dietz syndrome different from Marfan syndrome?
The two conditions share some features, including widening of the aorta and long, flexible limbs. However, LDS has certain features that Marfan syndrome does not typically show, such as widely spaced eyes, a split uvula or cleft palate, and twisted arteries. Conversely, dislocation of the lens of the eye, which occurs frequently in Marfan syndrome, does not happen in LDS. Importantly, tears in the aorta can occur at smaller sizes in LDS, which is why doctors recommend surgery earlier.
My child has been diagnosed with LDS but looks perfectly healthy — should I still be worried?
Yes, it is important to take the diagnosis seriously even if your child appears well on the outside. The most dangerous feature of LDS which is the widening and weakening of the arteries only shows up on scans. Some people with LDS have very few visible physical features but still carry significant vascular risks. Your child needs regular monitoring with echocardiograms and arterial imaging regardless of how they look.
What is the life expectancy for a child diagnosed with LDS today?
This is one of the most common questions families ask, and the answer is encouraging. Older studies suggested a shorter lifespan, but those figures came from the most severe cases, often diagnosed late. With early detection, modern medication, regular surveillance, and timely surgery, many people with LDS now live well into adulthood and beyond. The outlook continues to improve as treatments advance.
Can my child still play sports and be active?
Absolutely. Staying active matters. Swimming, cycling, walking, hiking, gentle jogging, and non-competitive racquet sports all work well. The key is to keep the intensity moderate: your child should feel able to talk comfortably during the activity. Families should avoid competitive and contact sports, heavy weight-lifting, and high-intensity exercises like push-ups and pull-ups that cause sudden spikes in blood pressure.
How often will my child need check-ups and scans?
Most children with LDS have an echocardiogram every 6 to 12 months and a full CT or MRI scan of the arteries from head to pelvis once a year. The exact frequency depends on the individual findings. It if the aorta stays stable and there are no other aneurysms, the intervals may stretch slightly. If the team detects rapid change, monitoring will happen more often.
Does my child need surgery, and when?
Not every child with LDS will need surgery, but many will at some point. Doctors recommend surgery when an aneurysm reaches a size where the risk of a tear becomes significant, typically around 4.0 cm in teenagers and adults, or earlier in younger children if the aorta is growing quickly. Surgeons most commonly perform a valve-sparing aortic root replacement, which has an excellent track record. The team always makes the decision about timing on an individual basis.
Should my other children and family members be tested?
Yes. If your child has received a diagnosis of LDS and testing has identified the specific gene change, parents and siblings should strongly consider genetic testing. About a quarter of cases come from a parent who may not even know they carry the gene change. Finding other affected family members early means they can start monitoring before any problems develop. This is one of the most important steps a family can take after a diagnosis.
Can Loeys-Dietz syndrome skip a generation?
In some of the rarer types of LDS (particularly types 4 and 5), a person can carry the gene change but show very few or no obvious signs of the condition. Their child, who inherits the same gene change, may then go on to develop more significant features. This can give the appearance of the condition skipping a generation. For this reason, even family members who seem unaffected should continue with imaging surveillance if testing has confirmed they carry the gene change.
Is there anything I should know in case of an emergency?
Sudden severe pain in the chest, back, or abdomen, especially if it feels like a tearing or ripping sensation, may signal an aortic dissection, which is a medical emergency. Call 999 immediately. Your child should carry a medical alert card or bracelet stating that they have Loeys-Dietz syndrome. Emergency teams need to know that the situation may require imaging of the full arterial tree (not just the heart), and that your child may take blood pressure medication. The Loeys-Dietz Syndrome Foundation provides downloadable emergency cards that you can keep in your child’s bag or wallet.
Dr. Alessandro Giardini, MD, PhD
Written 30/03/2026