Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) in Children: A Guide for Parents
By Dr Alessandro Giardini, Senior Consultant Paediatric and Adolescent Cardiologist, Great Ormond Street Hospital, London
Finding out that your child has been diagnosed with arrhythmogenic right ventricular cardiomyopathy, or that there is a family history of it, is naturally frightening. It is a condition with a serious sounding name and a genuine association with abnormal heart rhythms, but it is also one where early diagnosis, expert specialist care, and sensible lifestyle management can make an enormous difference to long term outcomes. This guide explains what ARVC is, what causes it, how it is investigated and treated in children and teenagers, and answers the questions that families most often bring to clinic with a paediatric cardiologist in London.
What is Arrhythmogenic Right Ventricular Cardiomyopathy?
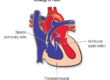
Arrhythmogenic right ventricular cardiomyopathy, ARVC, also previously known as arrhythmogenic right ventricular dysplasia, ARVD, is a condition in which the normal muscle tissue of the right ventricle, the chamber of the heart that pumps blood to the lungs, is progressively replaced by fatty and fibrous, scar tissue. This replacement process disrupts the normal electrical connections between heart muscle cells, causing the heart’s rhythm to become unstable and predisposing the affected person to potentially dangerous ventricular arrhythmias.
The right ventricle typically bears the greatest burden of the disease, but it is now well recognised that in a significant proportion of cases the left ventricle is also involved, either alongside the right, or occasionally as the dominant site. For this reason, the broader term “arrhythmogenic cardiomyopathy”, ACM, is increasingly used by specialists to reflect this wider spectrum of disease.
ARVC is a rare condition, estimated to affect approximately 1 in 2,000 to 1 in 5,000 people in the general population, though the true figure is likely higher because many cases remain undiagnosed. It is an important cause of sudden cardiac death in young people and in athletes, and it often runs in families.
What Causes ARVC?
ARVC is a genetic condition in the great majority of cases. The underlying defect lies in the genes that encode desmosomal proteins, the molecular “glue” that holds heart muscle cells together at points of contact. When these proteins are disrupted, heart muscle cells separate and die, particularly during times of mechanical stress such as exercise, and are replaced over time by fatty and fibrous tissue. The most frequently implicated genes include PKP2, plakophilin 2, DSP, desmoplakin, DSG2, DSC2, and JUP, though variants in a growing number of additional genes have been identified.

Most cases are inherited in an autosomal dominant pattern, meaning that a parent who carries the variant has a 50 percent chance of passing it on to each child. The condition may appear to skip generations because penetrance is variable, some individuals who carry a disease associated variant may have minimal or no detectable features, particularly in childhood.
The proportion of cases with a positive family history is substantial, though not universal. All first degree relatives, and in many cases second degree relatives, of someone with a confirmed diagnosis should be considered for specialist cardiac evaluation.
It is important to appreciate that carrying a gene variant associated with ARVC is not the same as having established disease. Many gene carriers, particularly younger children, have normal hearts on initial assessment. Disease expression tends to evolve over time, and the period of greatest risk and most active disease progression is typically in adolescence and young adulthood. This concealed phase of the condition is one reason why careful, ongoing monitoring of at risk family members is so important.
One additional factor of considerable practical significance is the relationship between vigorous exercise and ARVC. There is strong and consistent evidence that high intensity endurance exercise accelerates disease progression in genetically susceptible individuals and increases the risk of ventricular arrhythmia. This is one of the most important management considerations once a diagnosis is established or a pathogenic variant is identified.
What Are the Symptoms?
In children, ARVC can be an entirely silent condition, particularly in early or mild disease. The condition rarely presents before the age of ten, and even in affected teenagers, many have no symptoms until identified through family screening.
When symptoms do occur, the most common are palpitations, an awareness of rapid, irregular, or forceful heartbeats, and lightheadedness or fainting, particularly during or after exercise. These symptoms often reflect ventricular ectopic beats or episodes of ventricular tachycardia arising from the right ventricle. On an ECG, this type of tachycardia classically shows a left bundle branch block pattern, reflecting its right ventricular origin.
Chest discomfort can occur and, in some patients, may be associated with episodes that resemble myocarditis, so called “hot phases” of the disease, which are increasingly recognised as part of the ARVC and ACM spectrum. Breathlessness and fatigue, if present, tend to reflect more advanced disease with impairment of ventricular function.
In a small but important proportion of cases, the first manifestation of ARVC is sudden cardiac arrest, most commonly during or shortly after vigorous exercise. This is why the condition must be taken seriously when identified in a family, and why prompt, expert evaluation of all at risk relatives is essential.
How is ARVC Investigated?
The diagnosis of ARVC is not made from a single test. It is established using internationally accepted criteria, most recently refined in the Padua criteria, which combine findings from multiple investigations to classify the diagnosis as definite, borderline, or possible. Dr Giardini applies these criteria carefully in clinical practice, ensuring that each child is assessed in a structured and evidence based way.
The investigations routinely performed include a resting 12 lead ECG, which may show characteristic abnormalities including T wave inversion in the right precordial leads and, less commonly, an Epsilon wave, a small electrical deflection at the end of the QRS complex that is relatively specific but not always present.
Ambulatory ECG monitoring, Holter monitoring over 24 to 48 hours or longer, is used to detect ventricular ectopic beats and arrhythmias that may not be captured during a resting recording.
Cardiac MRI is the most informative imaging tool for ARVC. It provides detailed visualisation of ventricular structure and function, identifies regional wall motion abnormalities, and can detect areas of fibrosis using late gadolinium enhancement. Echocardiography remains important for assessing ventricular size and function and for longitudinal follow up.
Exercise testing provides useful information about arrhythmias provoked by exertion and helps guide activity advice. Signal averaged ECG is an additional specialised technique that may detect late potentials, although it is used less frequently in some centres.
Genetic testing is an important part of the evaluation and is typically offered to the affected child and their family. A positive genetic result allows cascade screening of relatives and helps identify those who require ongoing follow up. However, a negative genetic test does not exclude ARVC, and clinical evaluation remains essential.
What Treatments Are Available?
The overarching goals of treatment in ARVC are to reduce the risk of life threatening arrhythmias, preserve ventricular function, and allow children to live as normal a life as possible. There is currently no treatment that reverses the underlying disease process, but there are effective strategies to manage risk.
Activity restriction
This is one of the most important aspects of management. High intensity and competitive endurance sports are generally contraindicated in individuals with confirmed ARVC or a pathogenic gene variant, as exercise accelerates disease progression and increases arrhythmic risk. Dr Giardini discusses this in detail with families, aiming to strike a balance between safety and maintaining a healthy, active lifestyle. Moderate, recreational activity is usually appropriate.
Antiarrhythmic medication
Beta blockers are commonly used as first line therapy to reduce arrhythmic burden and blunt the heart rate response to exercise. Other medications such as sotalol or amiodarone may be considered in selected cases, with careful specialist supervision.
Catheter ablation
For children with recurrent ventricular tachycardia despite medical therapy, catheter ablation can reduce arrhythmia burden. It is important to recognise that ablation does not cure the underlying condition, and arrhythmias may recur over time.
ICD implantation
An implantable cardioverter defibrillator may be recommended in children at higher risk of life threatening arrhythmias, for example those with prior sustained ventricular tachycardia, previous cardiac arrest, or significant ventricular dysfunction. The decision is individualised and involves careful discussion with the family.
Heart failure management and transplantation
In more advanced cases, standard heart failure therapies are used. A small minority of patients may ultimately require cardiac transplantation.
Monitoring
Lifelong specialist follow up is essential. The frequency of review depends on the presence of symptoms, arrhythmias, or structural changes, as well as genetic status. Children who carry a gene variant but have no structural disease still require ongoing surveillance.
Frequently Asked Questions
How is ARVC different from other types of cardiomyopathy?
Unlike hypertrophic cardiomyopathy, HCM, which involves thickening of the heart muscle, or dilated cardiomyopathy, where the heart becomes enlarged and weakened, ARVC primarily affects the electrical stability of the heart due to fibrofatty replacement of the myocardium. Arrhythmias are often the earliest and most significant feature, although ventricular dysfunction may develop over time.
My child has no symptoms but carries the gene variant, do they need treatment?
Not necessarily treatment, but they do require expert follow up. A gene positive child with a normal heart does not usually need medication, but does need regular monitoring, clear activity guidance, and a personalised safety plan. Avoidance of high intensity competitive sport is a key early recommendation.
Is competitive sport really that dangerous in ARVC?
Yes. There is strong evidence that high intensity endurance exercise increases both disease progression and arrhythmic risk. This does not mean children should avoid all activity, far from it, but competitive endurance sport is generally not advisable in individuals with ARVC or pathogenic variants.
Should siblings and parents be tested?
Yes. ARVC is a familial condition, and first degree relatives should undergo cardiac evaluation. Where a genetic variant is identified, targeted testing can help distinguish those who require follow up from those who can be reassured.
What is the risk of sudden cardiac death in a child with ARVC?
The risk varies significantly between individuals. In many children, particularly early in the disease, the absolute risk is low. However, certain features, including prior sustained arrhythmias, ventricular dysfunction, and a strong family history, increase risk. Careful risk stratification and appropriate management significantly reduce this risk.
Can ARVC be cured?
There is currently no cure for ARVC. Management focuses on reducing arrhythmic risk, monitoring disease progression, and maintaining quality of life. With appropriate specialist care, many children live full and active lives.
My teenager is a keen athlete, can they ever return to sport?
This is often one of the most difficult discussions. In most cases of confirmed ARVC or pathogenic variants, return to high intensity competitive sport is not recommended. However, safe levels of activity can usually be identified, and Dr Giardini works closely with families to find a balanced and sustainable approach.
How often does my child need cardiology review?
This depends on whether your child has established disease or is an at risk relative. Gene positive children with normal findings are typically reviewed annually with ECG, Holter monitoring, and imaging. Those with established disease require more frequent follow up tailored to their condition.
Where can I see Dr Giardini for a private consultation?
Dr Giardini sees children and families in his private practice in London across several locations, including Great Ormond Street Hospital, The Portland Hospital, and Chase Lodge Hospital, and the Bupa Cromwell Hospital. Families can self refer or be referred by a GP or paediatrician.
Dr. Alessandro Giardini, MD, PhD
Written 02/04/2026